14.3.4 Some other gert commands
@@ -598,23 +615,23 @@14.3.4 Some other gert commandsgit_log()
git_push()## Write a new file, using here::here to specify the path
-writeLines("hello", here::here("R", "test-here.R"))
-
-## Another way is to use use_r
-usethis::use_r("test-file-github.R") # adds file to the project's R directory
-
-## For example, we might try adding something new
-gert::git_add("R/test-file-github.R")
-
-## Add commit of what was done
-gert::git_commit("uploaded test file")
-
-## Gives info about the commits
-gert::git_log()
-
-## Upload your changes from the local repo to GitHub
-gert::git_push() # IMPORTANT COMMAND
+## Write a new file, using here::here to specify the path
+writeLines("hello", here::here("R", "test-here.R"))
+
+## Another way is to use use_r
+usethis::use_r("test-file-github.R") # adds file to the project's R directory
+
+## For example, we might try adding something new
+gert::git_add("R/test-file-github.R")
+
+## Add commit of what was done
+gert::git_commit("uploaded test file")
+
+## Gives info about the commits
+gert::git_log()
+
+## Upload your changes from the local repo to GitHub
+gert::git_push() # IMPORTANT COMMAND
It might be more user-friendly to use the Git pane that appears in RStudio :)
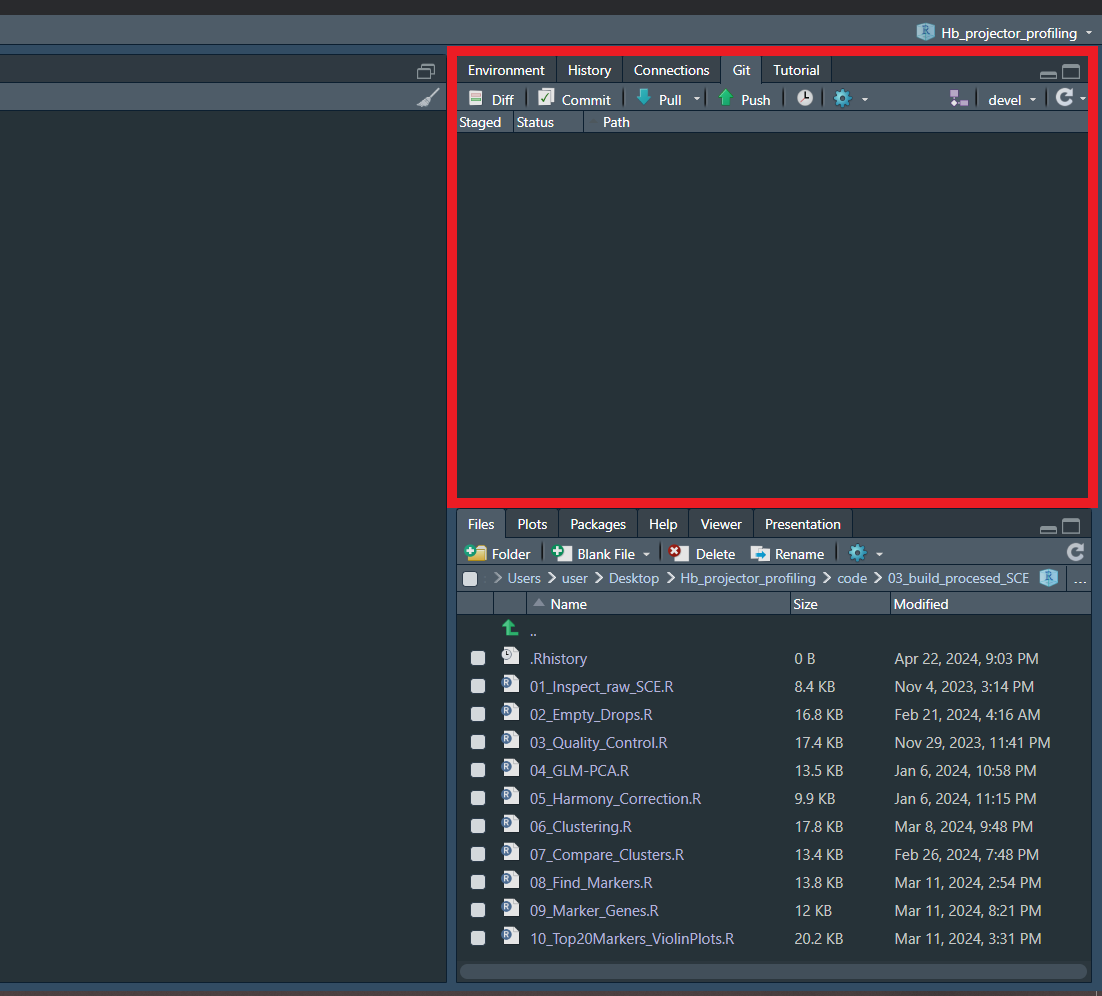
git_push()## Write a new file, using here::here to specify the path
-writeLines("hello", here::here("R", "test-here.R"))
-
-## Another way is to use use_r
-usethis::use_r("test-file-github.R") # adds file to the project's R directory
-
-## For example, we might try adding something new
-gert::git_add("R/test-file-github.R")
-
-## Add commit of what was done
-gert::git_commit("uploaded test file")
-
-## Gives info about the commits
-gert::git_log()
-
-## Upload your changes from the local repo to GitHub
-gert::git_push() # IMPORTANT COMMAND## Write a new file, using here::here to specify the path
+writeLines("hello", here::here("R", "test-here.R"))
+
+## Another way is to use use_r
+usethis::use_r("test-file-github.R") # adds file to the project's R directory
+
+## For example, we might try adding something new
+gert::git_add("R/test-file-github.R")
+
+## Add commit of what was done
+gert::git_commit("uploaded test file")
+
+## Gives info about the commits
+gert::git_log()
+
+## Upload your changes from the local repo to GitHub
+gert::git_push() # IMPORTANT COMMANDGit pane that appears in RStudio :)