Our tool is under active development and feedback is very much appreciated.
![]()
Multiple sequence alignment formulated as a machine learning problem, where an optimal profile hidden Markov model for a potentially ultra-large family of protein sequences is learned from unaligned sequences and an alignment is decoded. We use a novel, automatically differentiable variant of the forward algorithm to train pHMMs via gradient descent.
Since version 2.0.0, learnMSA can utilize protein language models (--use_language_model) for significantly improved accuracy.
- Aligns large numbers of protein sequences with state-of-the-art accuracy
- Can utilize protein language models (
--use_language_model) for significantly improved accuracy - Enables ultra-large alignment of millions of sequences
- GPU acceleration, multi-GPU support
- Scales linear in the number of sequences (does not require a guide tree)
- Memory efficient (depending on sequence length, aligning millions of sequences on a laptop is possible)
- Visualize a profile HMM or a sequence logo of the consensus motif
- Requires many sequences (in most cases starting at 1000, a few 100 might still be enough) to achieve state-of-the-art accuracy
- Only for protein sequences
- Increasingly slow for long proteins with a length > 1000 residues
learnMSA requires python <= 3.10 and TensorFlow >=2.5.0, <2.11. Version 2.10.* is recommended. Tensorflow should be installed with GPU support. We will try to support newer python and TensorFlow versions in the future.
Recommended way to install learnMSA along with Tensorflow + GPU:
- Create a conda environment:
conda create -n learnMSA python=3.10
conda activate learnMSA
- Install cuda toolkit in the environment:
conda install -c conda-forge cudatoolkit=11.2.2 cudnn=8.1.0
mkdir -p $CONDA_PREFIX/etc/conda/activate.d
echo 'export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:$CONDA_PREFIX/lib/' >> $CONDA_PREFIX/etc/conda/activate.d/env_vars.sh
conda deactivate
conda activate learnMSA
- Install learnMSA in the environment:
pip install learnMSA
conda install -c bioconda mmseqs2
- (optional) Verify that TensorFlow 2.10 and learnMSA are correctly installed:
python3 -c "import tensorflow as tf; print(tf.__version__, tf.config.list_physical_devices('GPU'))"
learnMSA -h
Another quick option is to use mamba (replace conda with mamba in above instructions). If you have problems to install some packages, set up Bioconda channels.
Recommended way to align proteins:
learnMSA -i INPUT_FILE -o OUTPUT_FILE --use_language_model --sequence_weights
Without language model support (faster):
learnMSA -i INPUT_FILE -o OUTPUT_FILE --sequence_weights
We always recommend to run learnMSA with the --sequence_weights flag to improve accuracy. This requires mmseqs2 to be installed. You can use conda for this: conda install -c bioconda mmseqs2. Sequence weights (and thus the mmseqs2 requirement) are turned off by default.
Since learnMSA version 1.2.0, insertions are aligned with famsa. This improves overall accuracy. The old behavior can be restored with the --unaligned_insertions flag.
To output a pdf with a sequence logo alongside the msa, use --logo. For a fun gif that visualizes the training process, you can use --logo_gif (attention, slows down training and should not be used for real alignments).
Run the notebooks learnMSA_demo.ipynb or learnMSA_with_language_model_demo.ipynb with juypter.
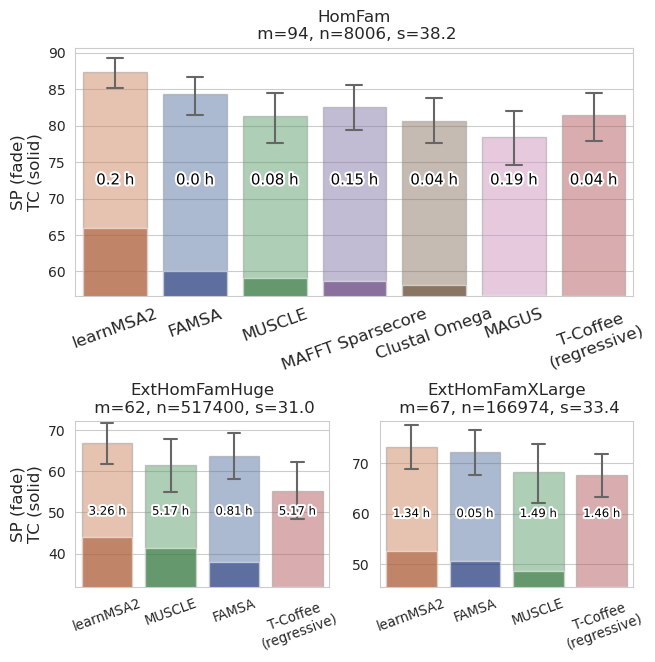
Becker F, Stanke M. learnMSA2: deep protein multiple alignments with large language and hidden Markov models. Bioinformatics. 2024
Becker F, Stanke M. learnMSA: learning and aligning large protein families. GigaScience. 2022