Pre
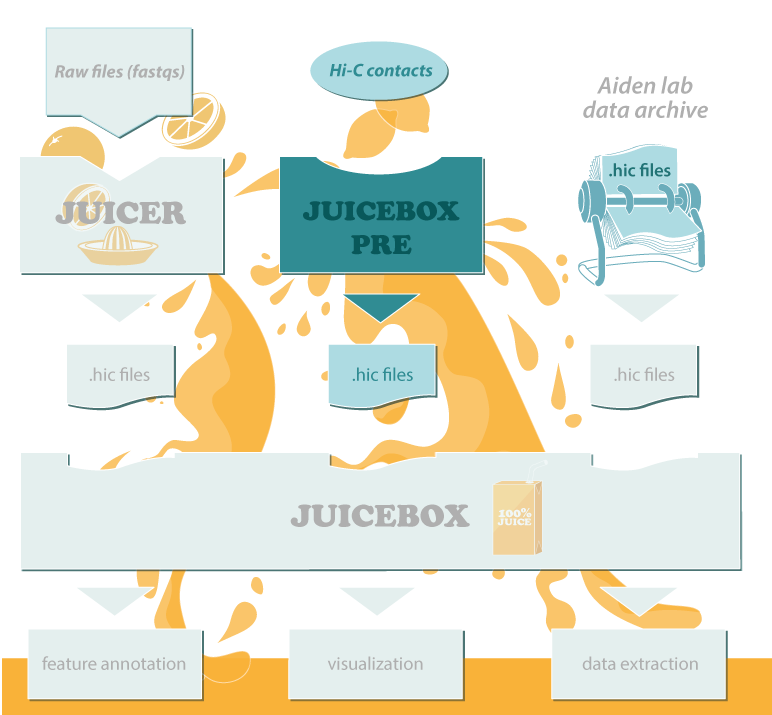 The Juicer Tools and Juicebox software is centered around the [*.hic* file](Data), a highly compressed binary file that provides rapid random access to the matrices. [*.hic* files](Data) can be visualized in the [Juicebox GUI](Visualization) and data can be easily extracted from them using the [*dump* command](Data-Extraction); our sophisticated [feature annotation algorithms](Feature-Annotation) also operate on .hic files.
The Juicer Tools and Juicebox software is centered around the [*.hic* file](Data), a highly compressed binary file that provides rapid random access to the matrices. [*.hic* files](Data) can be visualized in the [Juicebox GUI](Visualization) and data can be easily extracted from them using the [*dump* command](Data-Extraction); our sophisticated [feature annotation algorithms](Feature-Annotation) also operate on .hic files.
By default, .hic files are created from a text file describing the Hi-C reads and bin the reads at 9 base-pair-delimited resolutions: 2.5M, 1M, 500K, 250K, 100K, 50K, 25K, 10K, and 5K. If the user sends in a restriction site file, an additional 8 fragment-delimited resolutions are added: 500f, 250f, 100f, 50f, 20f, 5f, 2f, 1f.
The pre command allows users to create .hic files from their own data. It takes three required arguments and a number of optional arguments. The optional arguments should go before the required ones.
The required arguments are:
-
<infile>: Text file with paired contacts. The text file should be in one of the formats below. The text file may be gzipped. -
<outfile>: Name of outfile, should end with .hic. This is the file you will load into Juicebox or operate on using Juicebox tools. -
<genomeID>: Must be one of hg18, hg19, hg38, dMel, mm9, mm10, anasPlat1, bTaurus3, canFam3, equCab2, galGal4, Pf3D7, sacCer3, sCerS288c, susScr3, or TAIR10
The optional arguments are:
-
-dOnly calculate intra chromosome (diagonal) [false] -
-f <restriction site file>Calculate fragment map. Requires restriction site file; each line should start with the chromosome name followed by the position of each restriction site on that chromosome, in numeric order, and ending with the size of the chromosome [not set] -
-m <int>Only write cells with count above threshold m [0] -
-q <int>Filter by MAPQ score greater than or equal to q [not set] -
-c <chromosome ID>Only calculate map on specific chromosome [not set] -
-r <comma-separated lists of resolutions>Only calculate specific resolutions [not set] -
-t <tmpDir>Set a temporary directory for writing -
-s <statistics file>Add the text statistics file to the Hi-C file header -
-g <graphs file>Add the text graphs file to the Hi-C file header -
-nDon't normalize the matrices -
-vVerbose mode -
-hPrint help and exit
The pre command accepts several formats.
IMPORTANT NOTE pre throws away reads that map to the same restriction fragment. If you use dummy numbers for the frag field, be sure they are different for the different read ends; that is, <frag1> should be 0 and <frag2> should be 1.
A whitespace separated file that contains, on each line
<readname> <str1> <chr1> <pos1> <frag1> <str2> <chr2> <pos2> <frag2> <mapq1> <mapq2>
- str = strand (0 for forward, anything else for reverse)
- chr = chromosome (must be a chromosome in the genome)
- pos = position
- frag = restriction site fragment
- mapq = mapping quality score
If not using the restriction site file option, frag will be ignored, but please see above note on dummy values. If not using mapping quality filter, mapq will be ignored. readname and strand are also not currently stored within .hic files.
A whitespace separated file that contains, on each line
<str1> <chr1> <pos1> <frag1> <str2> <chr2> <pos2> <frag2>
- str = strand (0 for forward, anything else for reverse)
- chr = chromosome (must be a chromosome in the genome)
- pos = position
- frag = restriction site fragment
If not using the restriction site file option, frag will be ignored, but please see above note on dummy values. readname and strand are also not currently stored within .hic files.
This format is useful for reading in already processed files, e.g. those that have been already binned and/or normalized; this format can be easily used in conjunction with the -r flag to create a .hic file that contains a single resolution.
A whitespace separated file that contains, on each line
<str1> <chr1> <pos1> <frag1> <str2> <chr2> <pos2> <frag2> <score>
- str = strand (0 for forward, anything else for reverse)
- chr = chromosome (must be a chromosome in the genome)
- pos = position
- frag = restriction site fragment
- score = the score imputed to this read
If not using the restriction site file option, frag will be ignored, but please see above note on dummy values. readname and strand are also not currently stored within .hic files.
The long format is used by Juicer and takes in directly the merged_nodups.txt file.
A whitespace separated file that contains, on each line
<str1> <chr1> <pos1> <frag1> <str2> <chr2> <pos2> <frag2> <mapq1> <cigar1> <sequence1> <mapq2> <cigar2> <sequence2> <readname1> <readname2>
- str = strand (0 for forward, anything else for reverse)
- chr = chromosome (must be a chromosome in the genome)
- pos = position
- frag = restriction site fragment
- mapq = mapping quality score
- cigar = cigar string as reported by aligner
- sequence = DNA sequence
If not using the restriction site file option, frag will be ignored, but please see above note on dummy values. If not using mapping quality filter, mapq will be ignored. readname, strand, cigar, and sequence are also not currently stored within .hic files.
Data files used in these examples: test.txt.gz chrX_5KB.txt.gz
java -Xmx2g -jar juicebox_tools.jar pre data/test.txt.gz data/test.hic hg19
This will produce a file in data/test.hic that can be loaded into Juicebox. It will not include a fragment map and will not filter any of the reads.
java -Xmx2g -jar juicebox_tools.jar pre -q 10 data/test.txt.gz data/test.hic hg19
This will produce a file in data/test.hic that can be loaded into Juicebox. Reads with one or more end with MAPQ < 10 will not be included. It will not include a fragment map.
java -Xmx2g -jar juicebox_tools.jar pre -q 30 -f data/hg19_MboI.txt data/test.txt.gz data/test.hic hg19
This will produce a file in data/test.hic that can be loaded into Juicebox. Reads with one or more end with MAPQ < 30 will not be included. The restriction site file data/hg19_MboI.txt should list all of the restriction sites on the hg19 genome, where each line should start with the chromosome name followed by the position of each restriction site on that chromosome, in numeric order, and ending with the size of the chromosome. When loaded into Juicebox, users will be able to see the map at fragment resolution.
java -Xmx2g -jar juicebox_tools.jar pre -r 5000 -c X -v -d data/chrX_5KB.txt.gz data/chrX_5KB.hic hg19
This will produce a file in data/chrX_5KB.hic that can be loaded into Juicebox. Only chromosome X is examined and only 5Kb resolution is calculated. The -d flag indicates only intrachromosomal reads are examined. The reads in data/chrX_5KB.txt.gz are in the "short with score" format, having already been binned.
There are more examples in the benchmark script
The restriction file format is whitespace delimited and contains one line for each chromosome. The first field on each line is the chromosome name, followed by the locations of the restriction sites on that chromosome, in linear order. For example, the first few lines of hg19_DpnII.txt looks like this:
1 11160 12411 12461 ... 249250621
2 11514 11874 12160 ... 243199373
3 60138 60662 60788 ... 198022430
On chromosome 1, the DpnII cutting sequence GATC appears first at 11160, then at 12411, then at 12461, and so on. On chromosome 2, GATC appears first at 11514, then at 11874, then at 12160, etc. The last field is the size of the chromosome.
You can find some commonly used restriction site files on our Box mirror.
The Juicer script generate site positions creates this file from a given genome and restriction site.
TBA - information on how to create a properly formatted statistics and graphs file so that the "Dataset Metrics" tab in Juicebox will look normal. For now, see the statistics.pl file in Juicer for an example.
You can find the file specification for .hic files here and in the supplementary table to our Juicer paper. The [supplementary materials] (http://www.cell.com/cms/attachment/2065039642/2066196726/mmc1.pdf) also have further detail.