A computational toolkit in R for the integration, exploration, and analysis of high-dimensional single-cell cytometry data.
v0.4.1
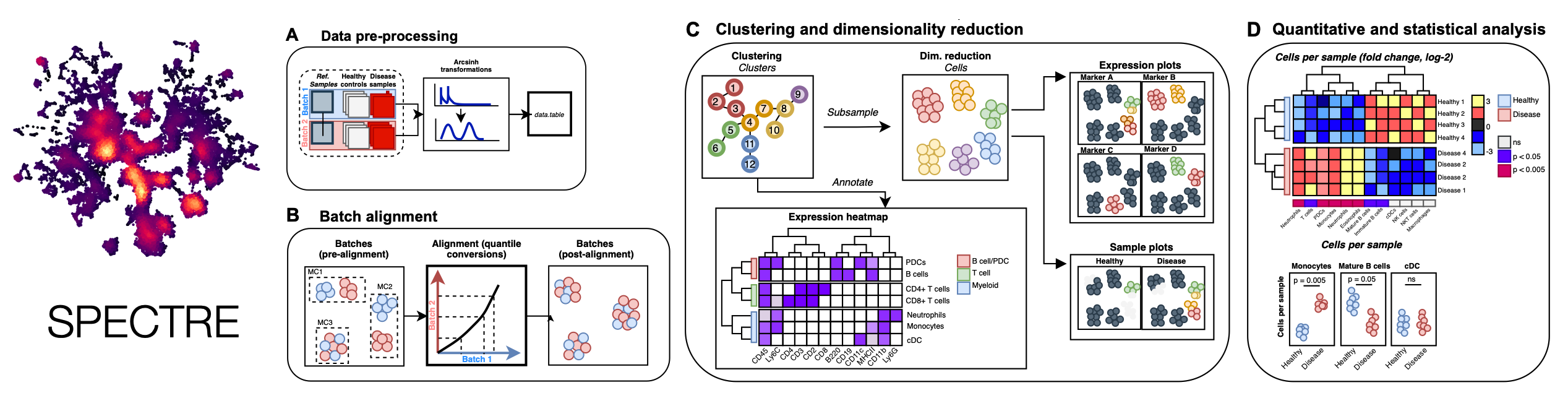
Spectre is an R package that enables comprehensive end-to-end integration and analysis of high-dimensional cytometry data from different batches or experiments. Spectre streamlines the analytical stages of raw data pre-processing, batch alignment, data integration, clustering, dimensionality reduction, visualisation and population labelling, as well as quantitative and statistical analysis. To manage large cytometry datasets, Spectre was built on the data.table framework – this simple table-like structure allows for fast and easy processing of large datasets in R. Critically, the design of Spectre allows for a simple, clear, and modular design of analysis workflows, that can be utilised by data and laboratory scientists. Recently we have extended the functionality of Spectre to support the analysis of Imaging Mass Cytometry (IMC) and scRNAseq data. For more information, please see our pre-print: Integration, exploration, and analysis of high-dimensional single-cell cytometry data using Spectre. (2020). TM Ashhurst*, F Marsh-Wakefield*, GH Putri*, AG Spiteri, D Shinko, ... bioRxiv.
Spectre was developed by Thomas Ashhurst, Felix Marsh-Wakefield, and Givanna Putri.
Usage instructions and protocols are available from https://wiki.centenary.org.au/display/SPECTRE.
If you use Spectre in your work, please consider citing Ashhurst TM, Marsh-Wakefield F, Putri GH et al. (2020). bioRxiv. 2020.10.22.349563. To continue providing open-source tools such as Spectre, it helps us if we can demonstrate that our efforts are contributing to analysis efforts in the community. Please also consider citing the authors of the individual packages or tools (e.g. CytoNorm, FlowSOM, tSNE, UMAP, etc) that are critical elements of your analysis work.
Detailed installation instructions are available from https://wiki.centenary.org.au/display/SPECTRE. Briefly, install and load the 'devtools' library.
if(!require('devtools')) {install.packages('devtools')}
library('devtools')
Subsequently, use the 'install_github' function to install and load the Spectre package. By default this will load the 'master' branch, which is the same as the latest stable release version (listed at https://github.com/sydneycytometry/Spectre/releases). To install a specific release version, see https://cran.r-project.org/web/packages/githubinstall/vignettes/githubinstall.html.
install_github("immunedynamics/spectre")
You will see the following returned:
Downloading GitHub repo sydneycytometry/spectre@master
These packages have more recent versions available.
Which would you like to update?
1: All
2: CRAN packages only
3: None
4: data.table (1.12.0 -> 1.12.2) [CRAN]
... etc
We suggest selecting 'none' (in this example, by entering '3' and pressing return) to avoid updating other packages.
If the package is sucessfully installed, you can load the library using:
library("Spectre")
Subsequently, there are a few packages from Bioconductor that you should install, which won't be installed by default when you install Spectre.
## Install BiocManager to download packages from Bioconductor
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
## Download additional BioConductor packages
if(!require('flowCore')) {BiocManager::install('flowCore')}
if(!require('Biobase')) {BiocManager::install('Biobase')}
if(!require('flowViz')) {BiocManager::install('flowViz')}
if(!require('FlowSOM')) {BiocManager::install('FlowSOM')}
You can then check for whether all of the packages for Spectre have been loaded correctly using the following commands
## Check if all required packages have been installed
Spectre::package.check()
## Load all required packages
Spectre::package.load()
Alternatively, you can go to releases (https://github.com/immunedynamics/spectre/releases) and download the latest stable release -- which can then be installed in R.
The Spectre package was constructed on the basis of the CAPX workflow in R (https://sydneycytometry.org.au/capx). Along with the various R packages used within Spectre, we would like to acknowledge the Seurat and cytofkit R packages from providing inspiration for elements of the package design.