![]()

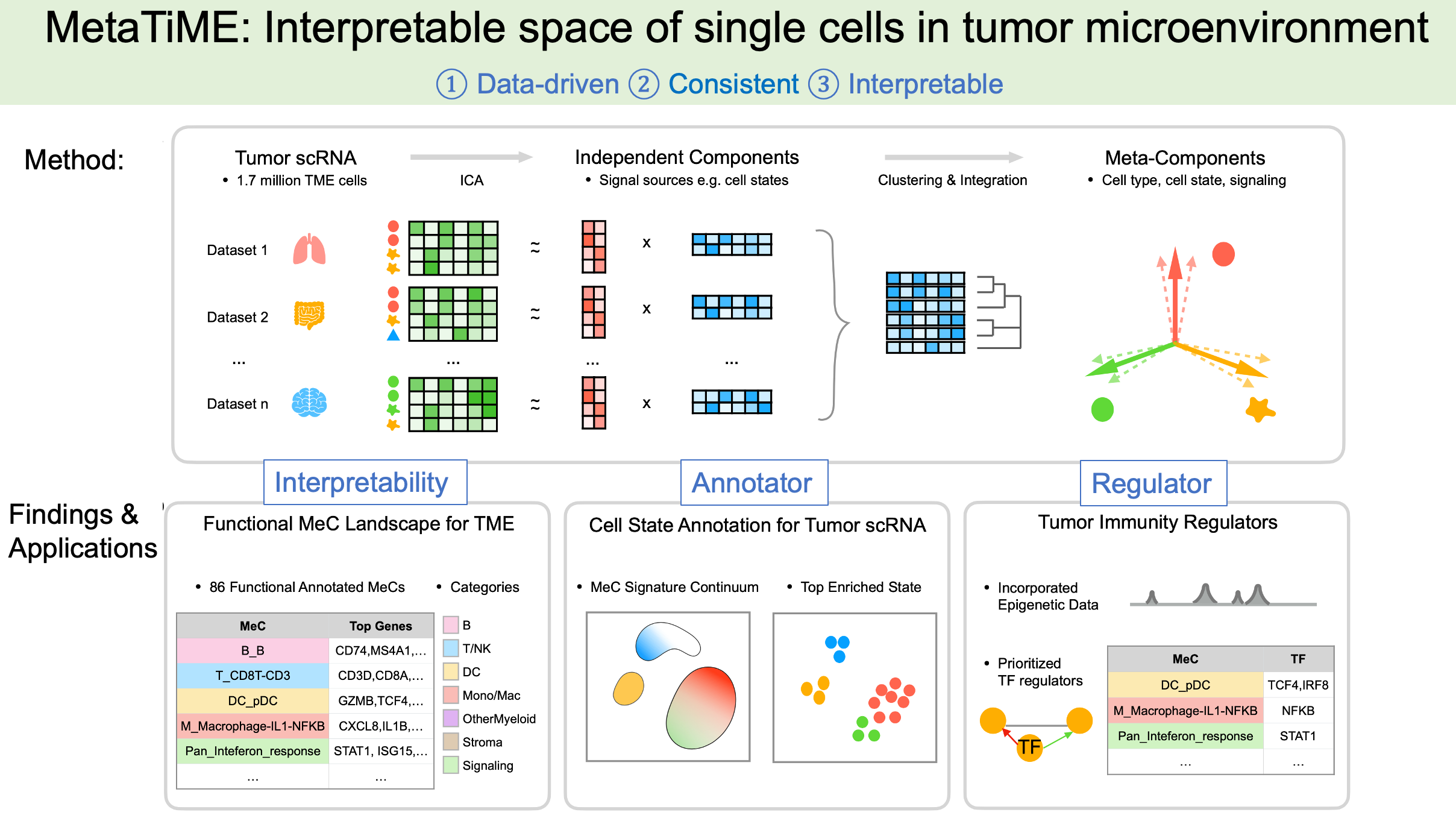
MetaTiME learns data-driven, interpretable, and reproducible gene programs by integrating millions of single cells from hundreds of tumor scRNA-seq data. The idea is to learn a map of single-cell space with biologically meaningful directions from large-scale data, which helps understand functional cell states and transfers knowledge to new data analysis. MetaTiME provides pretrained meta-components (MeCs) to automatically annotate fine-grained cell states and plot signature continuum for new single-cells of tumor microenvironment.
Create a new virtual env and activate (optional)
python -m venv metatime-env; source metatime-env/bin/activate
Use pip to install
pip install metatime
Installation shall be in minutes .
Next we have a tutorial on applying MetaTiME on new TME scRNAseq data to annotate cell states, scoring signature continuum, and test differential signature activity.
- Use MetaTiME to automatically annotate cell states and map signatures
- [New] MetaTiME re-training pipeline at https://github.com/yi-zhang/MetaTiMEpretrain/
Use MetaTiME to automatically annotate cell states and map signatures
Repo continously being improved! More details will be updated and suggested improvements welcome.
- [Paper at Nature Communications] (https://www.nature.com/articles/s41467-023-38333-8)
- Paper at bioRxiv
Tumor scRNAseq Data for MetaTiME @ Zenodo
-
A large collection of uniformly processed tumor single-cell RNA-seq.
-
Includes raw data and MetaTiME score for the TME cells.
- pandas
- scanpy
- anndata
- matplotlib
- adjustText
- leidenalg
- harmonypy
Dependency version tested:
- pandas==1.1.5
- scanpy==1.8.2
- anndata==0.8.0
- matplotlib==3.5.1
- adjustText==0.7.3
- leidenalg==0.8.3
Yi Zhang, Ph.D.
yiz [AT] ds.dfci.harvard.edu
Twitter | Website Research Fellow Department of Data Science Dana-Farber Cancer Institute Harvard University T.H. Chan School of Public Health